
Enhancer integration for network modeling
[grn In the intro, I described a three-part scheme to unravel the mystery of multicellular life. As part of that, I talked about how I mostly am trying to predict RNA levels these days. But, we know that important parts of the human regulatory network are contained in other types of molecules: for example, of all mutations related to autoimmunity, 90% are not in a coding region. This post discusses a class of gene-like entities called enhancers that have recently emerged as an interesting and potentially useful counterpart to genes. Teams are beginning to catalog enhancers and figure out how they help control cell state. This post will survey how that’s being done and will convey one important current question: how do we best connect each enhancer with the gene(s) it helps control?
How to measure noncoding DNA such as enhancers
Technologies for measuring the global state of DNA packaging (a.k.a. “chromatin”) are now quite mature. The technologies have Big Fancy Names such as:
- ChIP-seq: Chromatin Immuno-Precipitation followed by sequencing
- ATAC-seq: Assay for Transposase-Accessible Chromatin via sequencing
They use specially selected molecules that home in on interesting regions of DNA, enriching those regions for sequencing and leading to “peaks” of activity that can be displayed according to their location of the genome. (Examples below.) In this post, I’ll talk mostly about ATAC-seq.
What does ATAC-seq show?
ATAC-seq measures whether DNA is “open” enough for proteins to bind it, and it does this by sending in a protein called a transposase that can cut and mark those regions. Publicly or commercially available variants of that assay can now measure DNA accessibility in thousands of single cells, sometimes paired with RNA measurements or genetic perturbations. This post outlines ATAC-seq and how the results might be incorporated in network inference.

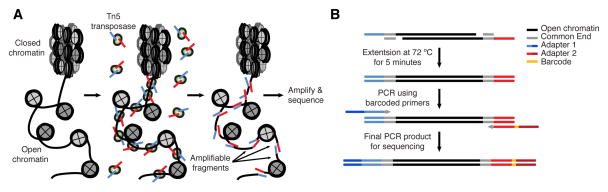
How ATAC-seq finds accessible DNA. This is from the original ATAC-seq paper, Buenrostro et al 2013.
The main output of ATAC-seq and related technologies is a set of “peaks”: spots on the genome with much higher signal than the surrounding areas. Here’s what this looks like (alongside competing technologies called FAIRE-seq and DNase-seq). The X axis is the position on the genome, and for each row, the height is the signal strength for each assay of DNA accessibility.


Signal peaks often occur at genes that are either transcriptionally active, or ready to become active. They also occur at small, discrete sites not located in genes, and these are often called enhancers. Enhancers are thought to serve as a landing pad for factors influencing transcription at nearby genes, and they are thought to contact genes in 3d, pinching off a loop of intervening DNA. There are millions of regions in the human genome that some people call enhancers.
Associating enhancers with the genes they regulate
To include enhancers in GRN inference, you have to somehow determine which gene each enhancer regulates. There are various ways of doing this.
- Perturbation: Some natural or CRISPR-induced genetic variants in enhancers can affect transcription. This is the topic of one of my all-time favorite studies, a highly multiplexed CRISPR enhancer screen by Gasperini and coauthors. The study found 664 high-confidence gene-enhancer pairs.
- Correlation: If enhancer A regulates gene B, then they should be correlated, and this requirement can pinpoint a few models that are compatible with the data. This is the idea behind CICERO, a single-cell ATAC analysis package built around its popular RNA counterpart, Monocle. Correlation-based linking of enhancers and genes is also used in a recent immgen consortium atlas of chromatin accessibility in the mouse immune system
- Proximity: Most enhancers regulate a nearby gene. According to that same ImmGen paper I mentioned above, the decay in rates of interaction is fast (exponential with genomic distance). In that study, many enhancers are within 50kb of their target gene (fig 3c), and very few are outside $5 \times 10^5$ basepairs (500kb). Likewise CICERO only bothers testing for links at a distance of $5 \times 10^5$ basepairs (500kb). According to Gasperini et al’s multiplexed CRISPR enhancer screen, the median distance across high-confidence gene-enhancer pairs is about $2.4 \times 10^4$ basepairs (24kb). The looping paper mentioned above finds that 27% of enhancers interact with the nearest TSS, and 47% of elements have interactions with the nearest expressed TSS. Some tools that need to connect enhancers with their targets simply take the nearest gene – for example, GREAT does this. This won’t work if you need a high fraction of correct pairings, but it’s OK in situations where you only need to enrich for correct answers.
- Looping: There are now many technologies that directly measure DNA looping, with names like 4C, 5C, Hi-C, Micro-C, and PLAC-seq. The general principle behind these techniques is to shear and re-ligate DNA while preserving its 3d structure. This yields fragments that are not contiguous in the linear genome sequence, but are nearby one another in 3d; sequencing can detect this as a way to infer 3d structure.
All of the techniques mentioned above are limited in different ways. Correlation-based analysis very promising, but doesn’t give clear answers about causality. Proximity-based pairing is only a heuristic; studies based on looping, correlation, and perturbation have all found that pairing each enhancer with the nearest gene is wrong most of the time (see below).
Perturbation-based pairing has a long way to go. CRISPR-perturbed single-cell RNA-seq is notoriously noisy, and the natural alternatives are limited. Gasperini et al’s screen sequenced 250k cells and found only 664 enhancer-gene pairs. There are hundreds of thousands of enhancers in the genome, so to scale up that tactic naively, we’d need to sequence hundreds of millions of cells. This may be affordable with some variant of the incredible MERFISH or hypr-seq, but we’re not there yet as of 2019.
DNA looping assays are an interesting case where you really need to know some quantities in order to determine what this technology can and cannot do. These datasets have very high background signal at short distances due to random polymer looping. Inside $10^6$ basepairs (1Mb), it wouldn’t be unusual to see interaction rates increase (very roughly) 10-fold with every 10-fold decrease in linear distance, e.g. in fig 1 here. Genome-wide, the overall enrichment is clear from the supplemental PDF (fig S10), but the noise makes it really hard to see individual enhancer-gene interactions, which often happen at the scale of 10^4 basepairs. Because of this limitation, the DNA looping literature is mostly concerned with big questions about principles of genome organization (such as during the cell cycle), rather than assigning specific regulatory elements to specific genes en masse. The one exception I have seen (so far) is this, which provides thousands of high-confidence gene-enhancer loops in table S6.
Even though enhancers don’t code for proteins, they are often transcribed into RNA. Recently, people have also started pairing enhancers and promoters via RNA-RNA contacts. The linked study finds about 10,000 enhancer-to-gene or gene-to-gene loops. For validation, they lowered enhancer RNA levels at 7 enhancers, and 27 of 31 paired genes had decreased transcription. Intervening on enhancer RNA’s is unusual – it would be more typical to use an enhancer trap (video) or a CRISPR intervention like Gasperini et al. do. It would be useful to validate a large simple random sample of RNA-RNA loops using a more conventional method, but so far this seems very promising.
For my interests, an enhancer is no use unless it can influence cell state, so the gold standard for gene-enhancer pairing is perturbation-based. Because of this, I judge each method by how well it can mimic perturbation data. Unfortunately, I know of no work assessing how well correlation-based approaches match perturbation-based enhancer-gene pairing. Correlation-based pairing is by far the easiest method to generate and analyze data for, so it would be great to know how well it can substitute for perturbation approaches.
What regulates enhancers?
To integrate a new class of elements into a GRN model, you need to know the outgoing links, the incoming links, and maybe some internal links. The section above details many approaches to inference of outgoing links: how enhancers regulate genes. Most of these are applicable to internal links as well (how enhancers regulate enhancers).
Unfortunately, it is more difficult to assess incoming links (how proteins regulate enhancers). This is really a topic for another post, but for now I have an overly brief explanation below.
There are technologies to measure incoming regulators directly: for example, ChIP-seq shows where on the genome a given protein binds. But, it requires additional data on top of what was just discussed, and it can only check one protein at a time. For yeast and fruit flies, most DNA binding proteins have been ChIP-seq’ed, but for humans and mice, we’re still missing a lot.
As a higher-throughput, low-quality alternative to ChIP-seq, ATAC-seq and DNase-seq data can be analyzed for recurring “motifs” in the DNA sequence where proteins tend to bind, but this type of analysis is very prone to false positives. The lowest false discovery rate I have ever seen when looking for the Foxn1 motif is about 0.4, meaning this motif is very difficult to distinguish from typical strings of nucleotides. Motif detection does not give clean enough results to confirm individual regulator-target pairs, so most practitioners clean up the signal either by averaging across many targets (as in TERA analysis) or by filtering with other types of data (as in SCENIC and CellOracle). Even if the motif is detected in a region of open chromatin, the corrresponding protein is not necessarily bound; there are other mechanisms to consider.
Also, just because a factor binds near a gene doesn’t mean it actually regulates transcription of that gene. To infer incoming arrows, motif analysis and ChIP-seq data could provide an initial boost, but I suspect the heavy lifting will have to be inferred from perturbation screens or observational data rather than direct binding.
Some references
- Cell Biology by the Numbers, http://book.bionumbers.org/how-many-mrnas-are-in-a-cell/
- Lineage tracing on transcriptional landscapes links state to fate during differentiation. Caleb Weinreb, Alejo E Rodriguez-Fraticelli, Fernando D Camargo, Allon M Klein. bioRxiv 467886; doi: https://doi.org/10.1101/467886
- Buenrostro, J. D., Wu, B., Chang, H. Y., & Greenleaf, W. J. (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Current protocols in molecular biology, 109, 21.29.1-9. doi:10.1002/0471142727.mb2129s109
- Chen, X., Miragaia, R. J., Natarajan, K. N., & Teichmann, S. A. (2018). A rapid and robust method for single cell chromatin accessibility profiling. Nature Communications, 9(1), 5345.
- 10X genomics application notes for single-cell ATAC-seq. https://www.10xgenomics.com/solutions/single-cell-atac/#application-notes
- Rubin, A. J., Parker, K. R., Satpathy, A. T., Qi, Y., Wu, B., Ong, A. J., … & Zarnegar, B. J. (2019). Coupled single-cell CRISPR screening and epigenomic profiling reveals causal gene regulatory networks. Cell, 176(1-2), 361-376. doi: 10.1016/j.cell.2018.11.022
- Cao, J., Cusanovich, D. A., Ramani, V., Aghamirzaie, D., Pliner, H. A., Hill, A. J., … & Steemers, F. J. (2018). Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science, 361(6409), 1380-1385.
- Duren, Z., Chen, X., Zamanighomi, M., Zeng, W., Satpathy, A. T., Chang, H. Y., … & Wong, W. H. (2018). Integrative analysis of single-cell genomics data by coupled nonnegative matrix factorizations. Proceedings of the National Academy of Sciences, 115(30), 7723-7728.
- Cusanovich, D. A., Reddington, J. P., Garfield, D. A., Daza, R. M., Aghamirzaie, D., Marco-Ferreres, R., … & Trapnell, C. (2018). The cis-regulatory dynamics of embryonic development at single-cell resolution. Nature, 555(7697), 538. PMID: PMC5866720. doi: 10.1038/nature25981.
- Gasperini, M., Hill, A. J., McFaline-Figueroa, J. L., Martin, B., Kim, S., Zhang, M. D., … & Trapnell, C. (2019). A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell, 176(1-2), 377-390.
- Pliner, H. A., Packer, J. S., McFaline-Figueroa, J. L., Cusanovich, D. A., Daza, R. M., Aghamirzaie, D., … & Adey, A. C. (2018). Cicero predicts cis-regulatory DNA Interactions from single-cell chromatin accessibility data. Molecular cell, 71(5), 858-871.
- Yoshida, H., Lareau, C. A., Ramirez, R. N., Rose, S. A., Maier, B., Wroblewska, A., … & Tellier, J. (2019). The cis-Regulatory Atlas of the Mouse Immune System. Cell. DOI:https://doi.org/10.1016/j.cell.2018.12.036
- Sanyal, A., Lajoie, B. R., Jain, G., & Dekker, J. (2012). The long-range interaction landscape of gene promoters. Nature, 489(7414), 109.